View Related Pages
Michael P. Sarras Jr., PhD
Cell Biology and Anatomy Discipline
Dr. Sarras received his BA in Biology from the University of Kansas, his MA in Cell and Developmental Biology from the University of Kansas, and his PhD from Louisiana State University Medical Center in Cell Biology and Anatomy. He went on to Postdoctoral Studies at Yale University School of Medicine in Cell Biology before accepting his first academic position at the University of Kansas, School of Medicine in 1981. In 1990 he completed an MBA from the University of Kansas, School of Business.
Over the course of his career, his research has focused on questions related to developmental and regenerative biology. Dr. Sarras served as an officer in the US Army during the Vietnam War and went to serve as an officer in the Research and Development Command until his retirement from the military in 1998 as a Lt. Col after 30 years service. At RFUMS he served as Vice President for Research and Dean of the Graduate School from 2003-2011. He retired from administrative duties in 2011 and currently holds the position of Professor of Cell Biology and Anatomy.
Research
Molecular mechanisms underlying the secondary complications of Diabetes Mellitus using Zebrafish as an experimental model in parallel with analysis of human diabetic tissues.
The results of several large scale diabetes mellitus (DM) clinical trials suggest that once initiated, diabetic complications persist and continue to progress unimpeded even when glycemic control is achieved through pharmaceutical intervention. This persistence has been supported by multiple lines of experimental laboratory evidence and collectively indicates that the initial hyperglycemic period results in permanent abnormalities in the target organs. This harmful phenomenon has been termed, metabolic memory (MM). The underlying molecular mechanisms of diabetic complications and metabolic memory may include the involvement of excess reactive oxygen species (ROS), the involvement of advanced glycation end products (AGE), and/or alterations in tissue-wide gene expression patterns. However, the ability to sustain these complications in the absence of hyperglycemia invokes a role for the epigenome in perpetuating these complications through metabolic memory.
The study of heritable changes in gene expression that cannot be explained by changes in the DNA sequence is referred to as epigenetics. There are several epigenetic processes that allow for altered gene expression including histone modification, de/methylation of cytosine residues within DNA, inclusion of histone variants in octomers, and control by non-coding RNAs. Altogether, these processes allow cells/organisms to quickly respond to changing environmental stimuli, and also confer the ability for the cell to "memorize" these encounters once the stimulus is removed. Recent in vitro investigations into the epigenome's role in metabolic memory have documented specific hyperglycemia induced histone modifications that persist in the post hyperglycemic (metabolic memory) state. Currently these reports have been somewhat limited to specific modifications or loci but it is expected that a more global approach will reveal a wealth of information.
Many roles have been ascribed to the presence of 5-methyl cytosine residues in DNA: gene silencing, silencing of transposable elements, developmental regulation of transcription, cell cycle control, differentiation, and recently the activation of genes. Not unexpectedly, due to the critical role in gene expression, aberrant DNA methylation has been associated with several human diseases including the susceptibility to diabetes mellitus or its complications. Additionally, the induction of tissue specific hypomethylation was reported in a diabetes mellitus type I rat model. Altered DNA methylation may become particularly important in the context of metabolic memory as only DNA methylation is backed by strong mechanistic support for heritability of epigenetic changes.
The long term objective of our laboratory is to decipher the molecular mechanisms of metabolic memory with a rationale that these studies will lead to the identification and development of novel therapeutic targets to control the progression of diabetic complications. To this end, we have developed an adult zebrafish model of type I diabetes mellitus using the diabetogenic drug, streptozocin. We have characterized this model to show that diabetic zebrafish not only display the known secondary complications including impaired epidermal wound healing, but in addition, exhibit impaired limb regeneration (caudal fin regeneration) as a consequence of diabetes mellitus induction. In our current studies we demonstrate that hyperglycemic zebrafish can revert back to normal glycemia within 2 weeks of drug removal due to regeneration of endogenous pancreatic beta cells resulting in a physiologically normal glycemic state. However, in contrast, body wall epidermal wound healing and limb regeneration in these fish remains impaired to the same extent as in the acute diabetic state indicating these complications are persistent and are susceptible to metabolic memory. Moreover, examination of daughter tissue that was regenerated in the post hyperglycemia state was similarly reduced revealing the heritable transmission of the metabolic memory phenomenon. In addition, we utilize the National Human Disease Tissue Bank to analyze what we learn from our zebrafish model in human diabetic tissues.
Zebrafish as a Developmental Model
Zebrafish has become a powerful developmental model because of its attributes of: 1) genetics, 2) rapid development (all systems essentially developed by 16 hours post-fertilization), 3) ex-utero development (ability to follow development on the bench top) 4) translucence (ability to visualized in real time, early and late phases of development), and 5) the wide variety of cellular and molecular techniques that can be applied to the system for functional analysis of development processes (transgenics, knockdown, and knock-out technologies).


Zebrafish as a Model for Regenerative Medicine
Zebrafish is one of the few vertebrates that can regenerate almost all of its organs and tissues in the adult state. The use of fin regeneration, for example, has been a powerful system to study the underlying mechanisms of regenerative biology. Experimentally, this also applies to a wide variety of other organs such as the 1) heart, 2) retina, 3) kidney, etc. The ability to induce regeneration and then follow it in a defined short time frame in combination with the developmental attributes described in the above paragraph has illuminated many of the underlying mechanisms of regeneration and has advanced the field of regenerative medicine.
Zebrafish as a Model of a Variety of Human Diseases.
Through transgenic and pharmaceutical approaches, a wide variety of human diseases have been induced in the zebrafish. This has allowed for detailed analysis of the underlying mechanisms of these diseases in this versatile animal model.
In 2010, our laboratory published a paper on development of an adult zebrafish model of Type I Diabetes (Ansgar Olsen, Michael P. Sarras, Jr., and Robert Intine. Limb regeneration is impaired in an adult zebrafish model of diabetes mellitus. Wound Repair and Regeneration. 2010, 18, 532-542).It was demonstrated in this model that IP injection of streptozotocin [STZ] (a pancreatic bela-cell toxin) induced a diabetic state in adult zebrafish (Fig 1) accompanied with the normal array of secondary complications that appeared within 4 weeks of the on-set of hyperglycemia (Figs 2 & 3).We also observed that fin regeneration (Fig. 4) was impaired in these diabetic fish (Fig. 5) which allowed us to then use this regenerative process to monitor the course of the disease in regard to problems related to regenerative medicine. Because of the high regenerative capacity of zebrafish, withdrawal of STZ was followed by a complete regeneration of pancreatic beta-cells and a return to normal glycemic levels. Despite a return to a condition of normal glycemic control, fin regeneration was still impaired and this condition was transmissible to new daughter cells as determined through repeated induction of fin regeneration in the now euglycemic zebrafish (Figs. 6 & 7). This means that the original state of hyperglycemia induced non-reversible alterations in the fin that manifested themselves as a permanent state of "impaired fin regeneration". Molecular analysis of fin cell's DNA found that in the new euglycemic state "metabolic memory" (continued tissue impairment after control of insulin levels have been achieved) alterations in the levels of DNA methylation had occurred. The laboratory is now investigating the molecular mechanism of hyperglycemia-induced DNA methylation in attempt to relate these DNA changes to impaired tissue function.
|
Figure 1
|
|
Figure 2
|
|
Figure 3
|
|
Figure 4
|
|
Figure 5
|
|
Figure 6 |
|
Figure 7 |
|
Figure 8 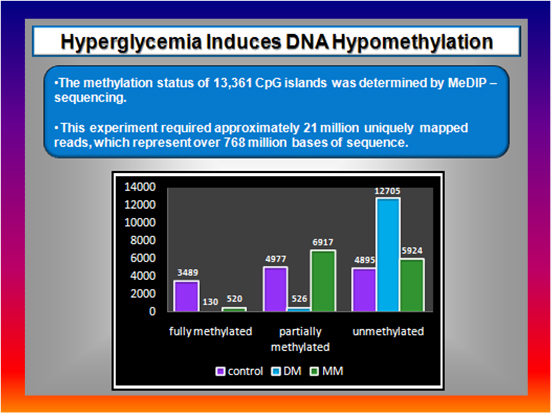 |







Publications
Articles published pertaining to current research program on the underlying mechanisms of Metabolic Memory in Diabetes that began in 2008 (of career total of 87 publications).
A. Selected Peer Reviewed Articles:
Ansgar Olsen, Michael P. Sarras, Jr., and Robert Intine. Impaired limb regeneration in a Zebrafish model of Diabetes. J. Wound Healing and Tissue Regeneration. 2010, 18(5):532-42.
Ansgar S. Olsen, Michael P. Sarras Jr., Alexey Leontovich, and Robert V. Intine. The heritable transmission of diabetic metabolic memory in zebrafish correlates with DNA hypomethylation and aberrant gene expression. Diabetes. 2012, 61(2),485-91.
Robert V. Intine, Ansgar S. Olsen, and Michel P. Sarras Jr. A zebrafish model of diabetes mellitus and metabolic memory. JOVE, 2013, 72 (e50232).
Michael. P. Sarras Jr.*, Alexey. A. Leontovich*, Ansgar. S. Olsen, and Robert. V. Intine. (*Co-first authors) Impaired tissue regeneration corresponds with altered expression of developmental genes that persists in the metabolic memory state of diabetic zebrafish. J. Wound Healing and Tissue Regeneration. 2013, 21(2), 320-328.
Pisano*, G., Mason*, S.M., Dhliway, N., Intine, R.V., Sarras, Michael Jr. (*Co-first authors) An Assay for Lateral Line Regeneration in Adult Zebrafish. JOVE, 2014, Apr 8;(86). doi: 10.3791/51343.
Dhliwayo, Nyembezi, Sarras, Michael Jr., Mason, Samantha, and Intine, Robert. Parp inhibition prevents ten eleven translocase enzyme activation and hyperglycemia induced DNA demethylation. Diabetes, 2014, 63:3069–3076. doi: 10.2337/db13-1916.
Michael P. Sarras Jr., Samantha Mason, Geoffrey McAllister, and Robert V. Intine. Inhibition of Poly-ADP Ribose Polymerase Enzyme Activity Prevents Hyperglycemia-Induced Impairment of Angiogenesis during Wound Healing. 2014, Sep;22(5):666-70.
Alexey A. Leontovich, Robert V. Intine, and Michael P. Sarras Jr., Epigenetic Studies point to DNA Replication/Repair Genes as a basis for the Heritable Nature of Long Term Complications in Diabetes. J. Diabetes Research, (In Press, 2016)
B. Invited Reviews:
Robert V. Intine and Michael P. Sarras Jr. Metabolic Memory and Chronic Diabetes Complications: Potential Role for Epigenetic Mechanisms. Curr. Diab. Rep. 12 (5):551-9, 2012.
Michael P. Sarras Jr. and Robert V. Intine. The Role of DNA Methylation in Type 1 and 2 Diabetes as Related to Endothelial Cell Dysfunction. Anatomy and Physiology, 1(1), 1-7, 2015.
Michael P. Sarras Jr. Editorial Overview of the Current Issue of Anatomy and Physiology. Anatomy and Physiology, 1(4), 1-2, 2015.
Michael. P. Sarras Jr, Alexey A. Leontovich, and Robert V. Intine. A Potential gDNA Methylation Epigenetic Mechanism to Explain the Long Term Complications of Type 1 and 2 Diabetes. Anat. and Physiology, 2016, In Press.
C. Invited Book Chapters:
Michael P. Sarras Jr. and Robert V Intine. Use of Zebrafish as a Disease Model Provides a Unique Window For Understanding the Molecular Basis of Diabetic Metabolic Memory. iConcept Press, 2013.